CROP-seq; 1:1:1 Mixture of DNMT3B, MBD1, and TET2 Knockout Cell Lines (HEK293T)¶
Dataset: CROP-seq; 1:1:1 Mixture of DNMT3B, MBD1, and TET2 Knockout Cell Lines (HEK293T)
Datlinger, P., Rendeiro, A.F., Schmidl, C., Krausgruber, T., Traxler, P., Klughammer, J., Schuster, L.C., Kuchler, A., Alpar, D., and Bock, C. (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301.
Preparation¶
Download fastq files from European Nucleotide Archive.
$ curl -O ftp.sra.ebi.ac.uk/vol1/fastq/SRR516/009/SRR5163029/SRR5163029_1.fastq.gz
$ curl -O ftp.sra.ebi.ac.uk/vol1/fastq/SRR516/009/SRR5163029/SRR5163029_2.fastq.gz
Mapping¶
This dataset is single-cell RNA-seq on HEK293T cell lines mixed in equal proportions knocked out for DNMT3B, MBD1, and TET2 (Fig. 1h). The platform of this dataset is Drop-seq. The details about the original data processing can be found here. Briefly, raw data were process with Drop-seq Tools v1.12 software. The first 12 bases on read 1 are cell barcodes, followed by 8 bases UMIs. Captured single cell transcripts are on read 2.
I re-processed these raw reads to get cell-associated barcodes for downstream analysis. Briefly, raw reads were trimmed using Trim Galore! and mapped to the human reference genome refdata-gex-GRCh38-2020-A using STAR v2.7.8a. Plasmid CROPseq-Guide-Puro sequence was not included in the reference.
Number of Read Pairs |
227,621,653 |
Number of Read Pairs, After Trimming |
201,527,916 |
Reads Mapped to Genome: Unique+Multiple |
0.7654 |
Reads Mapped to Genome: Unique |
0.696169 |
Estimated Number of Cells |
1,199 |
Fraction of Reads in Cells |
0.479281 |
Mean Reads per Cell |
45,290 |
Median Reads per Cell |
16,329 |
Mean UMI per Cell |
2,391 |
Median UMI per Cell |
1,118 |
Mean Genes per Cell |
1,042 |
Median Genes per Cell |
716 |
Total Genes Detected |
16,068 |
Inspect cell barcodes.
$ cat cell_barcodes.txt
AACGGGCATGGG
AACTGGGCATGG
AAGACAGCGTGT
AAGGGCGTACTC
AATAAATACAAA
AATAAATACAAC
AATAAATACAAG
AATAAATACAAT
AATCAATCGCAA
AATCAATCGCAC
Prepare feature barcodes. sgRNA sequences can be found in Supplementary Table 1.
$ cat feature_barcodes.tsv
DNMT3B CAGGATTGGGGGCGAGTCGG
MBD1 ATAGGTGTCTGAGCGTCCAC
TET2 CAGGACTCACACGACTATTC
Approach A¶
Since sgRNAs are captured through polyA tails together with other transcripts, the locations of sgRNA on read 2 vary (This is not a sgRNA enrichment library). To speed up processing, first we screen reads that have the constant sequence (GACGAAACACCG) upstream of sgRNAs (cutadapt, version 3.7).
$ cutadapt \
--cores 0 \
--front GACGAAACACCG \
--length 25 \
--minimum-length 25:25 \
--trimmed-only \
--output read_2_trimmed.fq.gz --paired-output read_1_trimmed.fq.gz \
../SRR5163029_2.fastq.gz ../SRR5163029_1.fastq.gz
Preview the filtering result: 1,429,437 out of 227,621,653 (0.6%) read pairs are kept for sgRNA identification.
== Read fate breakdown ==
Pairs that were too short: 25,972 (0.0%)
Pairs discarded as untrimmed: 226,166,244 (99.4%)
Pairs written (passing filters): 1,429,437 (0.6%)
QC¶
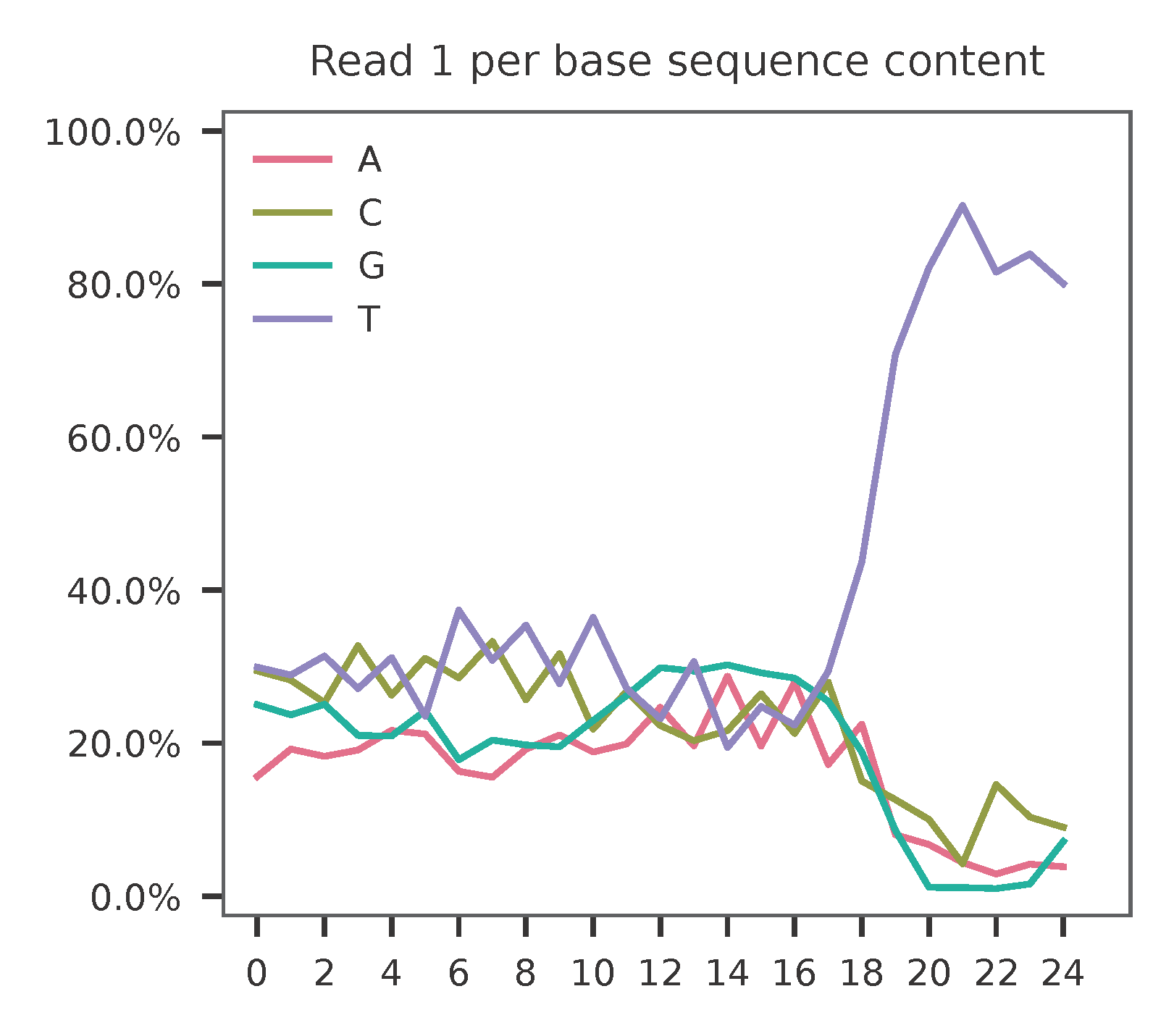
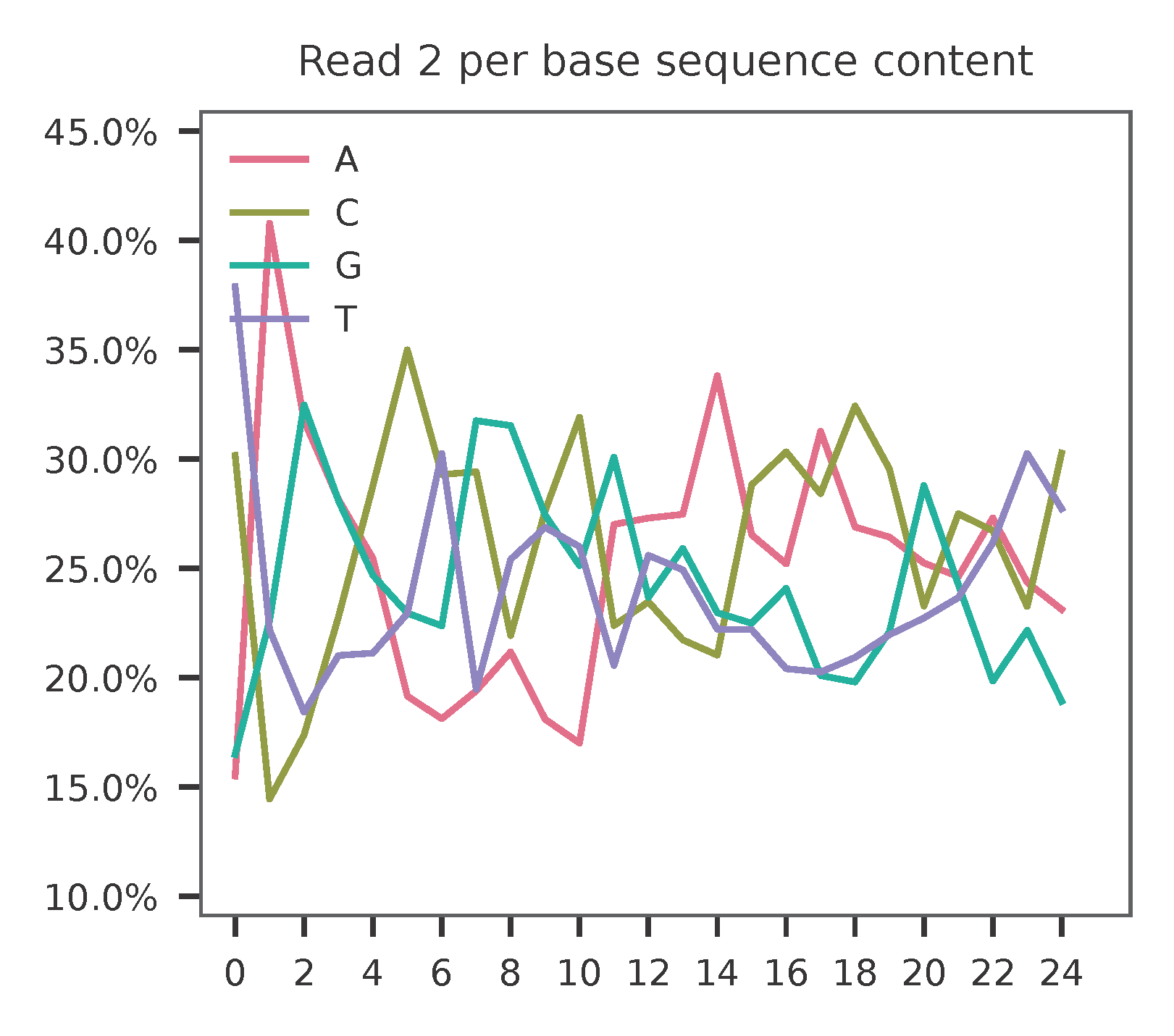
Sample the first 20,000 (set by -n, default 100,000) read pairs for quality control. Use -t to set the number of threads. By default, the diagnostic results and plots are generated in the qc directory (set by --output_directory), and full length of read 1 and read 2 are searched against reference cell and feature barcodes, respectively. The per base content of both read pairs and the distribution of matched barcode positions are summarized. Use -r1_c and/or -r2_c to limit the search range. Use -cb_n and/or -fb_n to set the mismatch tolerance for cell and feature barcode matching (default 3).
$ fba qc \
-1 read_1_trimmed.fq.gz \
-2 read_2_trimmed.fq.gz \
-w cell_barcodes.txt \
-f feature_barcodes.tsv \
-r1_c 0,12
This library is built using Drop-seq platform. The first 12 bases are cell barcodes and the following 8 bases are UMIs. Based on the base content plot, the GC content of cell barcodes are quite even. The UMIs are slightly T enriched.
As for read 2, the GC content of sgRNAs is quite even. The first 20 bases are sgRNA sequences.
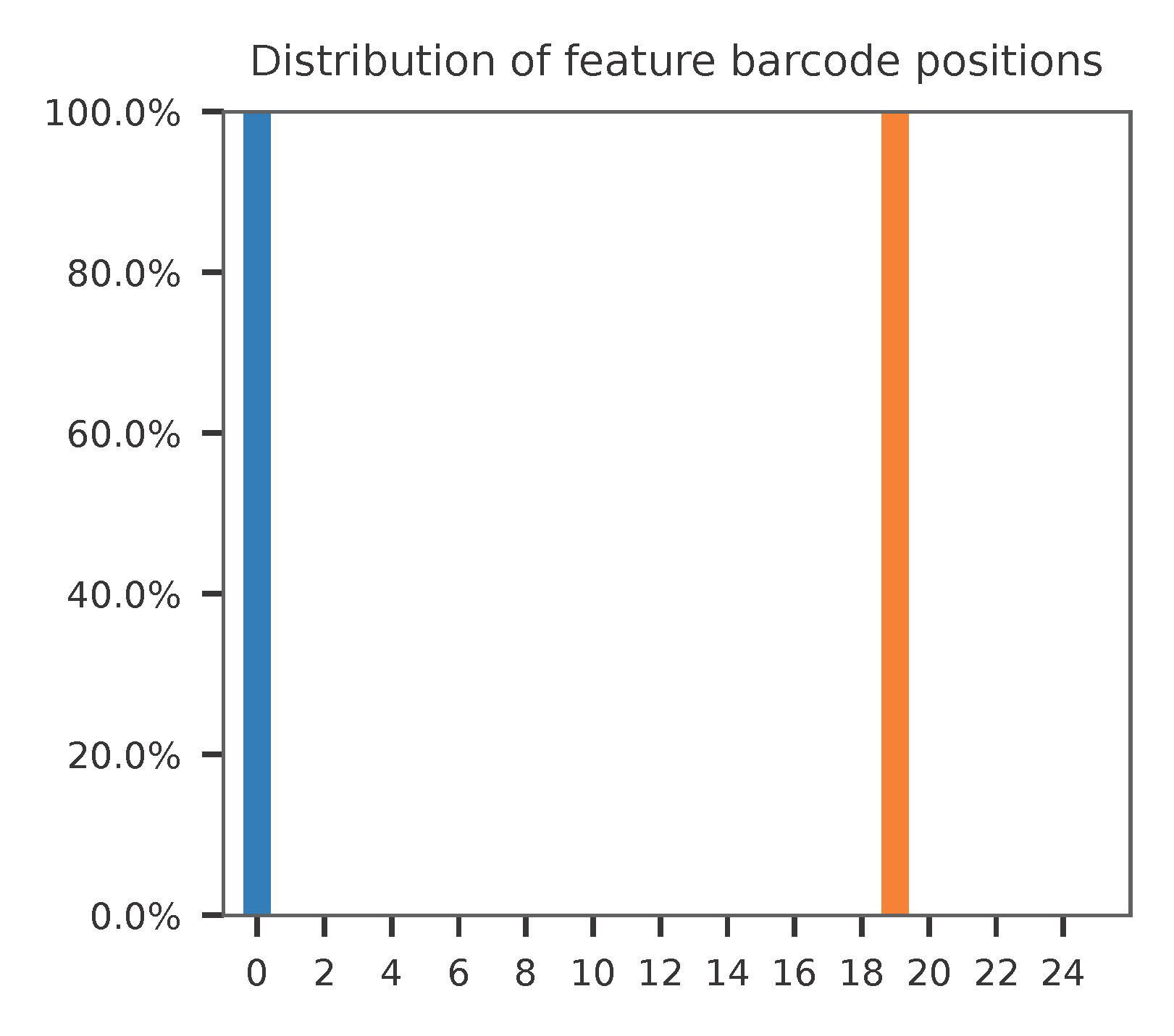
The detailed qc results are stored in feature_barcoding_output.tsv.gz file. matching_pos columns indicate the matched positions on reads. matching_description columns indicate mismatches in substitutions:insertions:deletions format.
$ zcat feature_barcoding_output.tsv.gz | grep -v no_match | head
read1_seq cell_barcode cb_matching_pos cb_matching_description read2_seq feature_barcode fb_matching_pos fb_matching_description
GCTGCATAGTCGggggggatttttt TTCATAGCTCCG 2:12 1:0:2 CAGGACTCACACGACTATTCGTTTT TET2_CAGGACTCACACGACTATTC 0:20 0:0:0
GTTGCTCCTCACggtgatttttttt GTTCCCTCCCAC 0:12 1:1:1 CAGGACTCACACGACTATTCGTTTT TET2_CAGGACTCACACGACTATTC 0:20 0:0:0
TAATGTTTAGGGagggcgctttttt TAATGTTTAGGG 0:12 0:0:0 ATAGGTGTCTGAGCGTCCACGTTTT MBD1_ATAGGTGTCTGAGCGTCCAC 0:20 0:0:0
TCTTCCACTACCggtatgacttttt TCTTCCACTACC 0:12 0:0:0 CAGGATTGGGGGCGAGTCGGGTTTT DNMT3B_CAGGATTGGGGGCGAGTCGG 0:20 0:0:0
GGAATGCCTTGAgtatacttttttt GGAATGCCTTGA 0:12 0:0:0 CAGGACTCACACGACTATTCGTTTT TET2_CAGGACTCACACGACTATTC 0:20 0:0:0
GCGATCACAATGtaatagatttttt GCGATCACAATG 0:12 0:0:0 CAGGATTGGGGGCGAGTCGGGTTTT DNMT3B_CAGGATTGGGGGCGAGTCGG 0:20 0:0:0
CGCCGTCGGACAcgaatcctttttt CCGTAGCGGGCA 2:12 1:0:2 ATAGGTGTCTGAGCGTCCACGTTTT MBD1_ATAGGTGTCTGAGCGTCCAC 0:20 0:0:0
CCGTCCTAGTTGatcccagtttttt CCGTCCTAGTTG 0:12 0:0:0 CAGGACTCACACGACTATTCGTTTT TET2_CAGGACTCACACGACTATTC 0:20 0:0:0
ATTGTTCCATCTgtcggcttttttt ACTGTTTGATCT 0:12 3:0:0 ATAGGTGTCTGAGCGTCCACGTTTT MBD1_ATAGGTGTCTGAGCGTCCAC 0:20 0:0:0
Barcode extraction¶
Search ranges are set to 0,12 on read 1 and 0,20 on read 2. One mismatch for cell and feature barcodes (-cb_m, -cf_m) are allowed.
$ fba extract \
-1 read_1_trimmed.fq.gz \
-2 read_2_trimmed.fq.gz \
-w cell_barcodes.txt \
-f feature_barcodes.tsv \
-o feature_barcoding_output.tsv.gz \
-r1_c 0,12 \
-r2_c 0,20 \
-cb_m 1 \
-fb_m 1
Preview of result.
$ gzip -dc feature_barcoding_output.tsv.gz | head
read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
TAATGTTTAGGGagggcgctttttt TAATGTTTAGGG 0 ATAGGTGTCTGAGCGTCCACgtttt MBD1_ATAGGTGTCTGAGCGTCCAC 0
TCTTCCACTACCggtatgacttttt TCTTCCACTACC 0 CAGGATTGGGGGCGAGTCGGgtttt DNMT3B_CAGGATTGGGGGCGAGTCGG 0
GGAATGCCTTGAgtatacttttttt GGAATGCCTTGA 0 CAGGACTCACACGACTATTCgtttt TET2_CAGGACTCACACGACTATTC 0
GCGATCACAATGtaatagatttttt GCGATCACAATG 0 CAGGATTGGGGGCGAGTCGGgtttt DNMT3B_CAGGATTGGGGGCGAGTCGG 0
CCGTCCTAGTTGatcccagtttttt CCGTCCTAGTTG 0 CAGGACTCACACGACTATTCgtttt TET2_CAGGACTCACACGACTATTC 0
ATTATATGTGAGcagactttttttt ATTATATGTGAG 0 ATAGGTGTCTGAGCGTCCACgtttt MBD1_ATAGGTGTCTGAGCGTCCAC 0
TTTCAGTATTGGggcgaattttttt TTTCAGTATTGG 0 ATAGGTGTCTGAGCGTCCACgtttt MBD1_ATAGGTGTCTGAGCGTCCAC 0
GTTCCCTCCCAAacatgagtttttt GTTCCCTCCCAA 0 CAGGATTGGGGGCGAGTCGGgtttt DNMT3B_CAGGATTGGGGGCGAGTCGG 0
GCTCCGCTTTTAactcaagtttttt GCTCCGCTTTTA 0 CAGGATTGGGGCCGAGTCGGgactt DNMT3B_CAGGATTGGGGGCGAGTCGG 1
Result summary.
9,213 out of 1,429,437 read pairs have valid cell and feature barcodes.
2022-03-07 16:11:53,295 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-07 16:11:53,295 - fba.__main__ - INFO - Initiating logging ...
2022-03-07 16:11:53,295 - fba.__main__ - INFO - Python version: 3.10
2022-03-07 16:11:53,295 - fba.__main__ - INFO - Using extract subcommand ...
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Number of reference cell barcodes: 1,199
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Number of reference feature barcodes: 3
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Read 1 coordinates to search: [0, 12)
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Read 2 coordinates to search: [0, 20)
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Cell barcode maximum number of mismatches: 1
2022-03-07 16:11:53,310 - fba.levenshtein - INFO - Feature barcode maximum number of mismatches: 1
2022-03-07 16:11:53,312 - fba.levenshtein - INFO - Read 1 maximum number of N allowed: 3
2022-03-07 16:11:53,312 - fba.levenshtein - INFO - Read 2 maximum number of N allowed: 3
2022-03-07 16:11:53,337 - fba.levenshtein - INFO - Matching ...
2022-03-07 16:12:13,951 - fba.levenshtein - INFO - Number of read pairs processed: 1,429,437
2022-03-07 16:12:13,952 - fba.levenshtein - INFO - Number of read pairs w/ valid barcodes: 9,213
2022-03-07 16:12:13,954 - fba.__main__ - INFO - Done.
Matrix generation¶
Only fragments with correct (passed the criteria) cell and feature barcodes are included. UMI removal is powered by UMI-tools (Smith, T., et al. 2017. Genome Res. 27, 491–499.). Use -us to set the UMI starting position on read 1 (default 16). Use -ul to set the UMI length (default 12). Fragments with UMI length less than this value are discarded. UMI deduplication method is set by -ud (default directional). Use -um to set UMI deduplication mismatch threshold (default 1).
The generated feature count matrix can be easily imported into well-established single cell analysis packages: Seruat and Scanpy.
$ fba count \
-i feature_barcoding_output.tsv.gz \
-o matrix_featurecount.csv.gz \
-us 12 \
-ul 8
Result summary.
2022-03-08 13:43:27,499 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-08 13:43:27,499 - fba.__main__ - INFO - Initiating logging ...
2022-03-08 13:43:27,499 - fba.__main__ - INFO - Python version: 3.9
2022-03-08 13:43:27,499 - fba.__main__ - INFO - Using count subcommand ...
2022-03-08 13:43:28,183 - fba.count - INFO - UMI-tools version: 1.1.1
2022-03-08 13:43:28,184 - fba.count - INFO - UMI starting position on read 1: 12
2022-03-08 13:43:28,184 - fba.count - INFO - UMI length: 8
2022-03-08 13:43:28,184 - fba.count - INFO - UMI-tools deduplication threshold: 1
2022-03-08 13:43:28,184 - fba.count - INFO - UMI-tools deduplication method: directional
2022-03-08 13:43:28,184 - fba.count - INFO - Header line: read1_seq cell_barcode cb_num_mismatches read2_seq feature_barcode fb_num_mismatches
2022-03-08 13:43:28,194 - fba.count - INFO - Number of lines processed: 9,213
2022-03-08 13:43:28,194 - fba.count - INFO - Number of cell barcodes detected: 420
2022-03-08 13:43:28,194 - fba.count - INFO - Number of features detected: 3
2022-03-08 13:43:28,194 - fba.count - INFO - UMI deduplicating ...
2022-03-08 13:43:28,202 - fba.count - INFO - Total UMIs after deduplication: 1,089
2022-03-08 13:43:28,202 - fba.count - INFO - Median number of UMIs per cell: 1.0
2022-03-08 13:43:28,204 - fba.__main__ - INFO - Done.
Demultiplexing¶
Gaussian mixture model¶
The implementation of demultiplexing method 2 (set by -dm) is inspired by the method described on 10x Genomics’ website. Use -p to set the probability threshold for demulitplexing (default 0.9). Use -nc to set the number of positive cells for a feature to be included for demultiplexing (default 200).
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-dm 2 \
-v \
-nc 0
2022-03-07 19:57:14,925 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-07 19:57:14,925 - fba.__main__ - INFO - Initiating logging ...
2022-03-07 19:57:14,925 - fba.__main__ - INFO - Python version: 3.9
2022-03-07 19:57:14,925 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-07 19:57:17,564 - fba.__main__ - INFO - Skipping arguments: "-q/--quantile", "-cm/--clustering_method"
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - Output directory: demultiplexed_gm
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - Demultiplexing method: 2
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - Visualization: On
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-07 19:57:17,564 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-07 19:57:17,571 - fba.demultiplex - INFO - Number of cells: 420
2022-03-07 19:57:17,571 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 0
2022-03-07 19:57:17,572 - fba.demultiplex - INFO - Number of features: 3 / 3 (after filtering / original in the matrix)
2022-03-07 19:57:17,572 - fba.demultiplex - INFO - Features: DNMT3B MBD1 TET2
2022-03-07 19:57:17,572 - fba.demultiplex - INFO - Total UMIs: 1,081 / 1,081
2022-03-07 19:57:17,573 - fba.demultiplex - INFO - Median number of UMIs per cell: 1.0 / 1.0
2022-03-07 19:57:17,573 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-07 19:57:18,277 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-07 19:57:18,423 - fba.demultiplex - INFO - Embedding ...
2022-03-07 19:57:21,922 - fba.__main__ - INFO - Done.
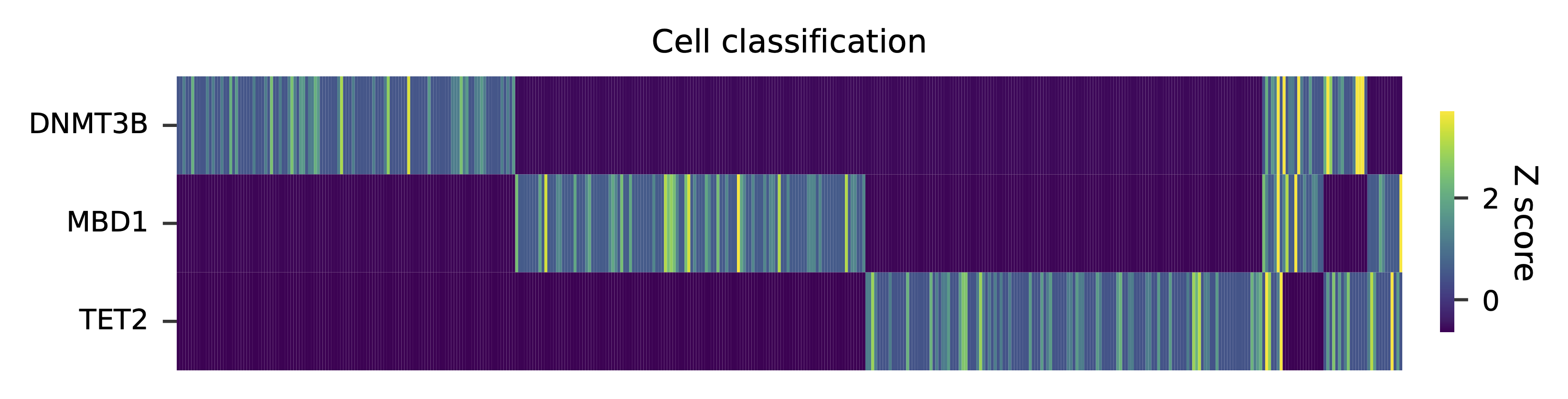
Heatmap of the relative abundance of features (sgRNAs) across all cells. Each column represents a single cell.
Preview the demultiplexing result: the numbers of singlets and multiplets.
In [1]: import pandas as pd
In [2]: m = pd.read_csv("demultiplexed/matrix_cell_identity.csv.gz", index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
DNMT3B 141
MBD1 150
TET2 158
dtype: int64
In [4]: sum(m.sum(axis=0) > 1)
Out[4]: 74
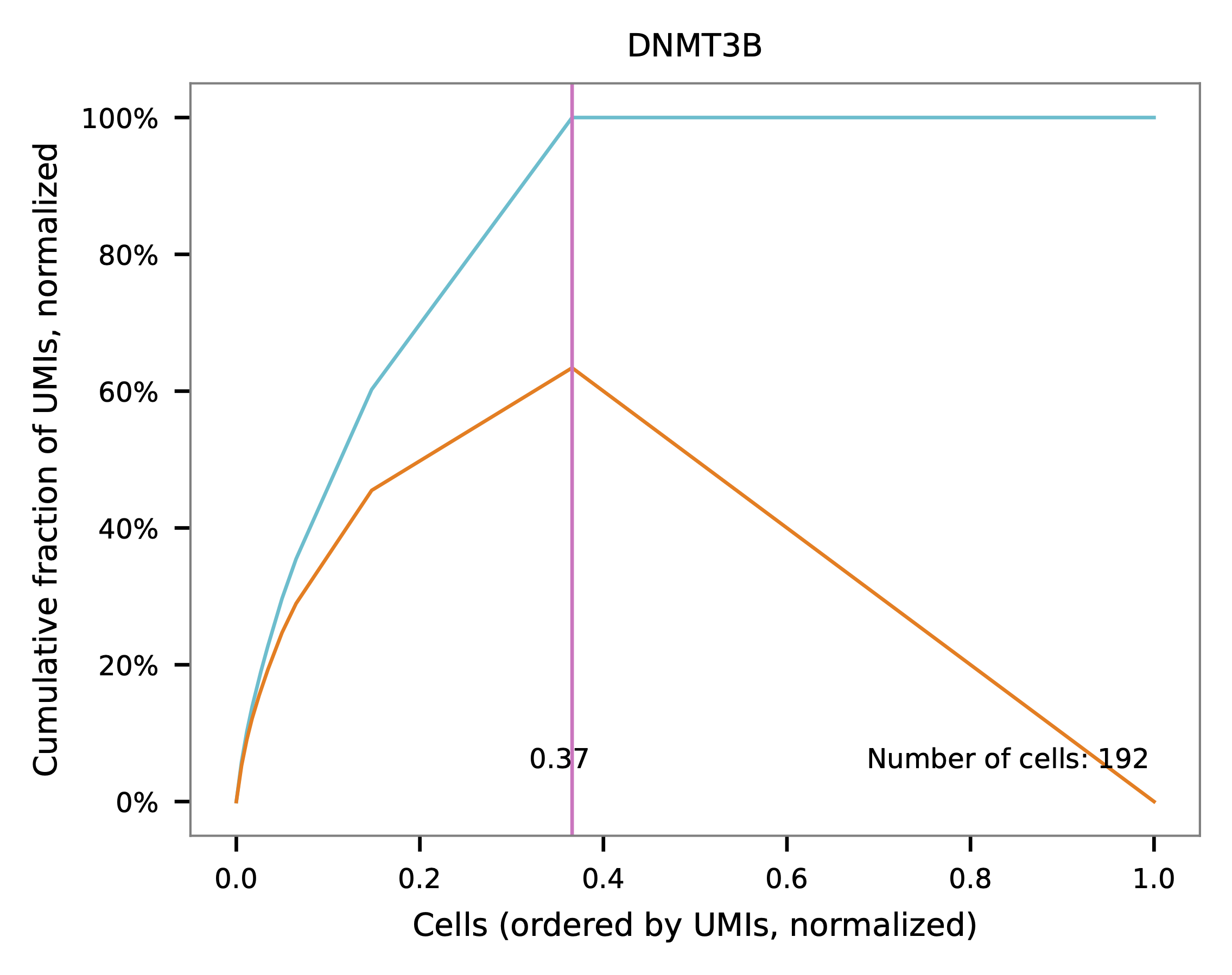
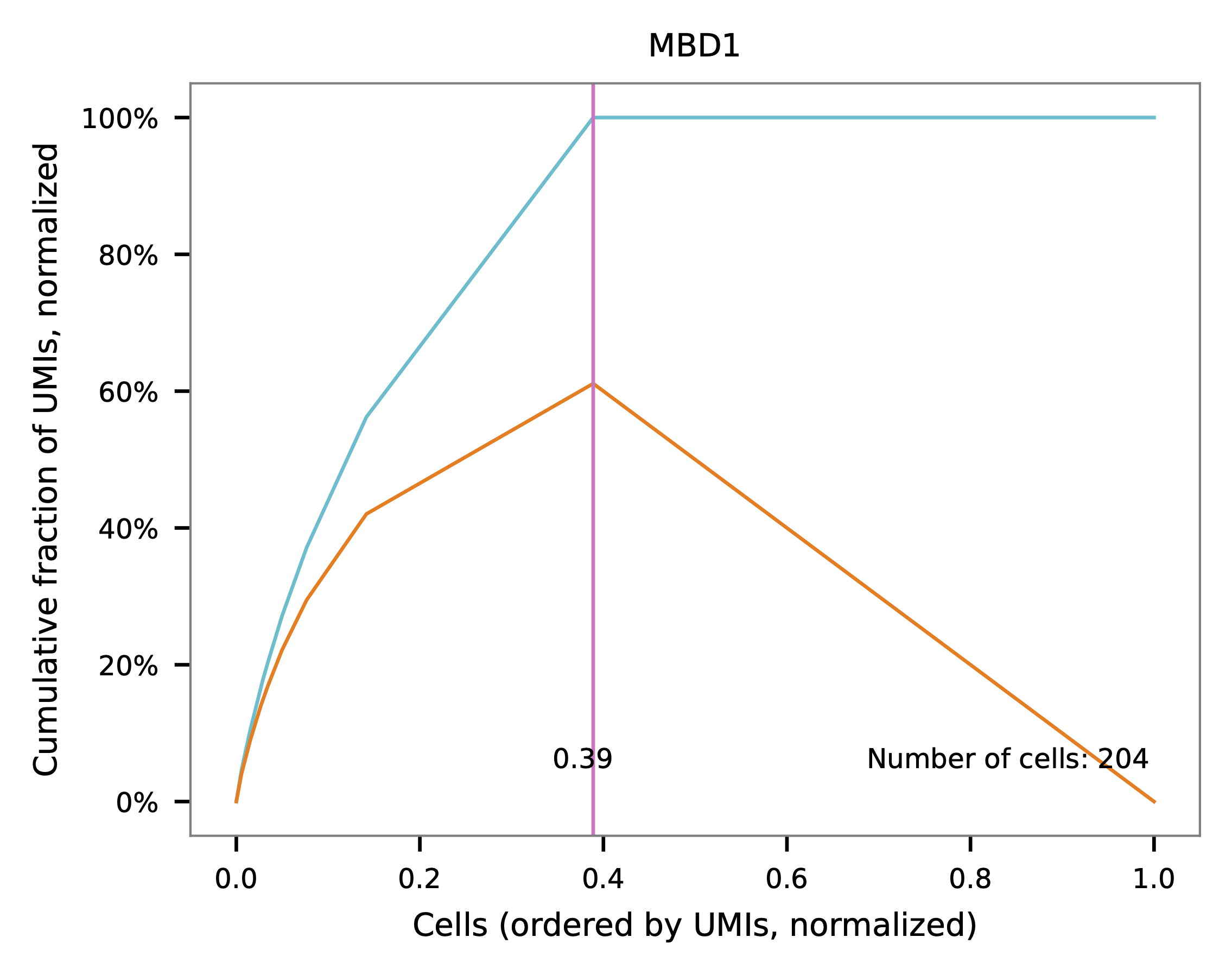
Knee point¶
Cells are demultiplexed based on the abundance of features (sgRNAs). Demultiplexing method 5 is implemented to use the local maxima on the difference curve to detemine the knee point on the UMI saturation curve.
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-dm 5 \
-v \
-nc 0
2022-03-05 01:52:38,900 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Initiating logging ...
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Python version: 3.9
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-05 01:52:41,396 - fba.__main__ - INFO - Skipping arguments: "-q/--quantile", "-cm/--clustering_method", "-p/--prob"
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Output directory: demultiplexed
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Demultiplexing method: 5
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Visualization: On
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-05 01:52:41,403 - fba.demultiplex - INFO - Number of cells: 523
2022-03-05 01:52:41,403 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 0
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Number of features: 3 / 3 (after filtering / original in the matrix)
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Features: DNMT3B MBD1 TET2
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Total UMIs: 1,364 / 1,364
2022-03-05 01:52:41,405 - fba.demultiplex - INFO - Median number of UMIs per cell: 1.0 / 1.0
2022-03-05 01:52:41,405 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-05 01:52:41,810 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-05 01:52:41,979 - fba.demultiplex - INFO - Embedding ...
2022-03-05 01:52:44,840 - fba.__main__ - INFO - Done.
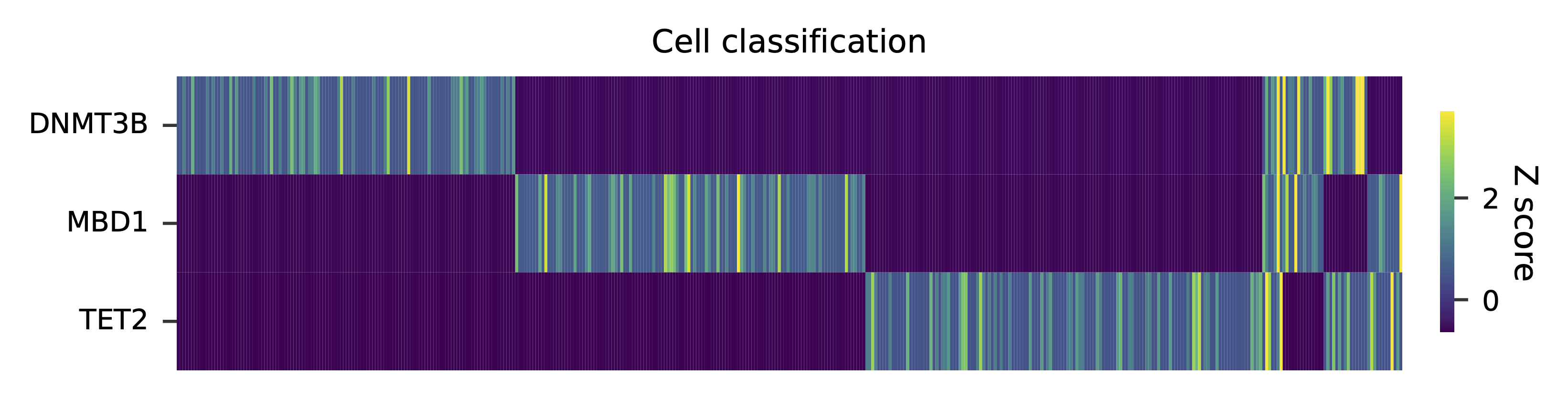
Heatmap of the relative abundance of features (sgRNAs) across all cells. Each column represents a single cell.
Preview the demultiplexing result: the numbers of singlets and multiplets.
In [1]: import pandas as pd
In [2]: m = pd.read_csv('demultiplexed/matrix_cell_identity.csv.gz', index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
DNMT3B 141
MBD1 150
TET2 158
dtype: int64
In [4]: sum(m.sum(axis=0) > 1)
Out[4]: 74
Approach B¶
Instead of pre-filtering read 2 for the constant upstream region of sgRNA, we search sgRNAs across the whole read 2. This mode is relatively slow, it is recommended to split fastq files and run on different nodes simultaneously to speed up.
Barcode extraction¶
The transcripts derived from CROPseq-Guide-Puro and captured by Drop-seq beads contain sgRNA sequences. There are no secondary libraries built on top of this single-cell RNA-seq library for sgRNA enrichment. The transcripts derived from CROPseq-Guide-Puro are captured by the ployA tails. Therefore, the locations of sgRNA on read 2 vary. We need to extract the sgRNA sequences from read 2.
qc mode is used for sgRNA extraction. Use -n to specify the number of reads to analyze, None means all the reads. Use -t to set the number of threads. By default, the diagnostic results and plots are generated in the qc directory (set by --output_directory), and full length of read 1 and read 2 are searched against reference cell and feature barcodes, respectively. The per base content of both read pairs and the distribution of matched barcode positions are summarized. Use -r1_c and/or -r2_c to limit the search range for read 1 and read 2 respectively. Use -cb_n and/or -fb_n to set the mismatch tolerance for cell and feature barcode matching (default 3).
$ fba qc \
-1 SRR5163029_1.fastq.gz \
-2 SRR5163029_2.fastq.gz \
-w cell_barcodes.txt \
-f feature_barcodes.tsv \
-cb_m 1 \
-fb_m 1 \
-cb_n 15 \
-fb_n 15 \
-r1_c 0,12 \
-t $SLURM_CPUS_ON_NODE \
--num_reads None
The detailed qc results are stored in feature_barcoding_output.tsv.gz file. matching_pos columns indicate the matched positions on reads. matching_description columns indicate mismatches in substitutions:insertions:deletions format.
$ gzip -dc qc/feature_barcoding_output.tsv.gz | head
read1_seq cell_barcode cb_matching_pos cb_matching_description read2_seq feature_barcode fb_matching_pos fb_matching_description
TTTAGGATCGTTtgatgtattttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttcttctttcttttttattctttacaacatcctaccataacata no_match NA NA ATTAAAAATATTGTGGCAGGAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACAAAAAAAAACAAAAAAAAATCAGCTATATAACCACTAATACTTCTA NA NA NA
GTCGAAACTCTTaacgggatttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttttt no_match NA NA TTATAATGGTTACAAATAAAGCAATAGCATCACAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA NA NA NA
GTTTACGTGTTCatgggcgattttttttttttttttttttttttttttttaaaaaagttaaaagggggcccgtggggggacaaatagaggggcctagagttccaccccccatcccacaaaaaaaaccctcaccgcacagggcctcgcccct GTTTACGTGTTC 0:12 0:0:0 GGAGTACGGAGAATTCTATAAGAGCTTGACCAATGACTGGGAAGATCACTTGGCAGTGAAGCATTTTTCAGTTGAAGGACAGTTGGAATTCAGAGCCCTTCTATTTGTCCCACGACGTGCTCCTTTTGATCTGTTTGAAAAAAAAAAAAAA no_match NA NA
CCGTCCTAGTTGgtgtatattttttttgtttttttttttttttcaccgggtcagagctgcccctaagtaccacgtcccgtcccacctttatcggacctcggccaccacaaattgcttatccagagtgcccccctccgcccatcccagactc CCGTCCTAGTTG 0:12 0:0:0 AATTAAGTCTCGTAAAGAACGAGAAGCTGAACTTGGACCTAGGGCAACCGACTTCACCAATGTTTACAGCGAGAATCTTGGTGACGACGTGGATGATGAGCGCCTTAAGGTTCTCTTTGGCAAGTTTGGGCCTGCCTTGAGTGTGCGACTT no_match NA NA
TTTCAGTATTGGggcgaattttttttttttttttttttttttttttttttttttttttttttttggctagtttttttgtggtttttgcttttggttctctcgtttgccctggagctcccaggtccctttcttgtcctaccataggtaaccc TTTCAGTATTGG 0:12 0:0:0 GGACGAAACACCGATAGGTGTCTGAGCGTCCACGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTAAGCTTGGCGTAACTAGATCTTGAGACACTGCTTTT MBD1_ATAGGTGTCTGAGCGTCCAC 13:33 0:0:0
CTAGGTACCACTagacagtttttttttttttttttttttttttttttttttttttttttttctctatgtgtgcttttttttggctttagtctgtgggtccctagttagccccggcgcccccacgcgcagaacgtgtcttaccacaagaacc CTAGGTACCACT 0:12 0:0:0 TTCTTGGGTAGTTTGCAGTTTTTAAAATTATGTTTTAAAATGGACTATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAACGGACGAAACACCGATAGGTGTCTGAGCGTCCACGTTTTAGAGC MBD1_ATAGGTGTCTGAGCGTCCAC 121:1410:0:0
TCTTCCACTACCgtcccgtcttttttttttttttttttttttttttttttttttttttctttatgtcagttttttttgtgctttagtattgggttcccttgtttgcccgagggctcccaggcccagatttgggctaaccaaagggaccccg TCTTCCACTACC 0:12 0:0:0 ACCGATAGGTGTCTGAGCGTCCACGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGAGTCGGTGCTTTTTTAAGCTTGGCGTAACTAGATCTTGAGACACTGCTTTTTGCTTGTAC MBD1_ATAGGTGTCTGAGCGTCCAC 4:24 0:0:0
CTTAATTTGGTGggaagattttttttttttttttttttttttttttttaagtactttaagtaagctttttttaggctttagccgtgggttcccctgttagcccgggaggtccccgggcccaatctgggcctaacagagaggccccgtacaa CTTAATTTGGTG 0:12 0:0:0 CCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGCAGGACTCACACGACTCTTCGTTTTAGAGCTAGCAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAAAAGTGGCACCGT TET2_CAGGACTCACACGACTATTC 63:83 1:0:0
TCGTACATACGGtggtttttttttttttttttttttttttttttttttttttttttttttttttttgtttttttttttttttgtttttttttttgtgtcctttgttttcactggggctcccaggtccatatccggtgttaccagagaaacc TCGTACATACGG 0:12 0:0:0 ATCATATGCTTACCGTAACTTGAAAGTATTTCGATTTCTTGGCTTTATATATCTTGTGGAAAGGACGAAACACCGCAGGATTGGGGGCGAGTCGGGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAGGCTAGTCCGTTATCAACTTGAAA DNMT3B_CAGGATTGGGGGCGAGTCGG 75:95 0:0:0
Matrix generation¶
Only fragments with correct (passed the criteria) cell and feature barcodes are included. UMI removal is powered by UMI-tools (Smith, T., et al. 2017. Genome Res. 27, 491–499.). Use -us to set the UMI starting position on read 1 (default 16). Use -ul to set the UMI length (default 12). Fragments with UMI length less than this value are discarded. UMI deduplication method is set by -ud (default directional). Use -um to set UMI deduplication mismatch threshold (default 1).
The generated feature count matrix can be easily imported into well-established single cell analysis packages: Seruat and Scanpy.
$ fba count \
-i feature_barcoding_output.tsv.gz \
-o matrix_featurecount.csv.gz \
-us 12 \
-ul 8
Result summary.
11.76 % (1,364 out of 11,597) of read pairs with valid cell and feature barcodes are unique fragments.
2022-03-04 23:18:27,501 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-04 23:18:27,501 - fba.__main__ - INFO - Initiating logging ...
2022-03-04 23:18:27,501 - fba.__main__ - INFO - Python version: 3.10
2022-03-04 23:18:27,501 - fba.__main__ - INFO - Using count subcommand ...
2022-03-04 23:18:31,494 - fba.count - INFO - UMI-tools version: 1.1.2
2022-03-04 23:18:31,495 - fba.count - INFO - UMI start position on read 1 auto-detected, overriding -us
2022-03-04 23:18:31,495 - fba.count - INFO - UMI length: 8
2022-03-04 23:18:31,496 - fba.count - INFO - UMI-tools deduplication threshold: 1
2022-03-04 23:18:31,496 - fba.count - INFO - UMI-tools deduplication method: directional
2022-03-04 23:18:31,496 - fba.count - INFO - Header line: read1_seq cell_barcode cb_matching_pos cb_matching_description read2_seq feature_barcode fb_matching_pos fb_matching_description
2022-03-04 23:18:31,581 - fba.count - INFO - Number of lines processed: 11,597
2022-03-04 23:18:31,581 - fba.count - INFO - Number of cell barcodes detected: 523
2022-03-04 23:18:31,582 - fba.count - INFO - Number of features detected: 3
2022-03-04 23:18:31,608 - fba.count - INFO - Total UMIs after deduplication: 1,364
2022-03-04 23:18:31,609 - fba.count - INFO - Median number of UMIs per cell: 1.0
2022-03-04 23:18:31,615 - fba.__main__ - INFO - Done.
Demultiplexing¶
Gaussian mixture model¶
The implementation of demultiplexing method 2 (set by -dm) is inspired by the method described on 10x Genomics’ website. Use -p to set the probability threshold for demulitplexing (default 0.9). Use -nc to set the number of positive cells for a feature to be included for demultiplexing (default 200).
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-dm 2 \
-v \
-nc 0
2022-03-04 23:19:05,218 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-04 23:19:05,219 - fba.__main__ - INFO - Initiating logging ...
2022-03-04 23:19:05,219 - fba.__main__ - INFO - Python version: 3.10
2022-03-04 23:19:05,219 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-04 23:19:15,199 - fba.__main__ - INFO - Skipping arguments: "-q/--quantile", "-cm/--clustering_method"
2022-03-04 23:19:15,200 - fba.demultiplex - INFO - Output directory: demultiplexed
2022-03-04 23:19:15,201 - fba.demultiplex - INFO - Demultiplexing method: 2
2022-03-04 23:19:15,201 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-04 23:19:15,201 - fba.demultiplex - INFO - Visualization: On
2022-03-04 23:19:15,201 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-04 23:19:15,201 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-04 23:19:15,219 - fba.demultiplex - INFO - Number of cells: 523
2022-03-04 23:19:15,219 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 0
2022-03-04 23:19:15,222 - fba.demultiplex - INFO - Number of features: 3 / 3 (after filtering / original in the matrix)
2022-03-04 23:19:15,222 - fba.demultiplex - INFO - Features: DNMT3B MBD1 TET2
2022-03-04 23:19:15,222 - fba.demultiplex - INFO - Total UMIs: 1,364 / 1,364
2022-03-04 23:19:15,223 - fba.demultiplex - INFO - Median number of UMIs per cell: 1.0 / 1.0
2022-03-04 23:19:15,223 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-04 23:19:17,319 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-04 23:19:17,784 - fba.demultiplex - INFO - Embedding ...
2022-03-04 23:19:32,256 - fba.__main__ - INFO - Done.
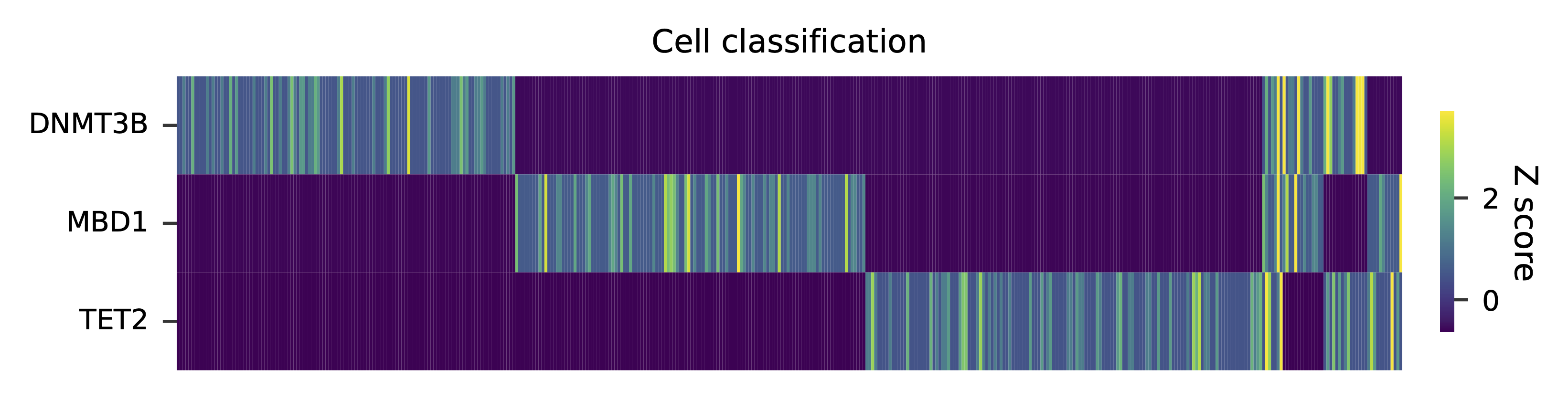
Heatmap of the relative abundance of features (sgRNAs) across all cells. Each column represents a single cell.
Preview the demultiplexing result: the numbers of singlets and multiplets.
In [1]: import pandas as pd
In [2]: m = pd.read_csv("demultiplexed/matrix_cell_identity.csv.gz", index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
DNMT3B 141
MBD1 150
TET2 158
dtype: int64
In [4]: sum(m.sum(axis=0) > 1)
Out[4]: 74
Knee point¶
Cells are demultiplexed based on the abundance of features (sgRNAs). Demultiplexing method 5 is implemented to use the local maxima on the difference curve to detemine the knee point on the UMI saturation curve.
$ fba demultiplex \
-i matrix_featurecount.csv.gz \
-dm 5 \
-v \
-nc 0
2022-03-05 01:52:38,900 - fba.__main__ - INFO - fba version: 0.0.x
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Initiating logging ...
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Python version: 3.9
2022-03-05 01:52:38,900 - fba.__main__ - INFO - Using demultiplex subcommand ...
2022-03-05 01:52:41,396 - fba.__main__ - INFO - Skipping arguments: "-q/--quantile", "-cm/--clustering_method", "-p/--prob"
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Output directory: demultiplexed
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Demultiplexing method: 5
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - UMI normalization method: clr
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Visualization: On
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Visualization method: tsne
2022-03-05 01:52:41,396 - fba.demultiplex - INFO - Loading feature count matrix: matrix_featurecount.csv.gz ...
2022-03-05 01:52:41,403 - fba.demultiplex - INFO - Number of cells: 523
2022-03-05 01:52:41,403 - fba.demultiplex - INFO - Number of positive cells for a feature to be included: 0
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Number of features: 3 / 3 (after filtering / original in the matrix)
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Features: DNMT3B MBD1 TET2
2022-03-05 01:52:41,404 - fba.demultiplex - INFO - Total UMIs: 1,364 / 1,364
2022-03-05 01:52:41,405 - fba.demultiplex - INFO - Median number of UMIs per cell: 1.0 / 1.0
2022-03-05 01:52:41,405 - fba.demultiplex - INFO - Demultiplexing ...
2022-03-05 01:52:41,810 - fba.demultiplex - INFO - Generating heatmap ...
2022-03-05 01:52:41,979 - fba.demultiplex - INFO - Embedding ...
2022-03-05 01:52:44,840 - fba.__main__ - INFO - Done.
Heatmap of the relative abundance of features (sgRNAs) across all cells. Each column represents a single cell.
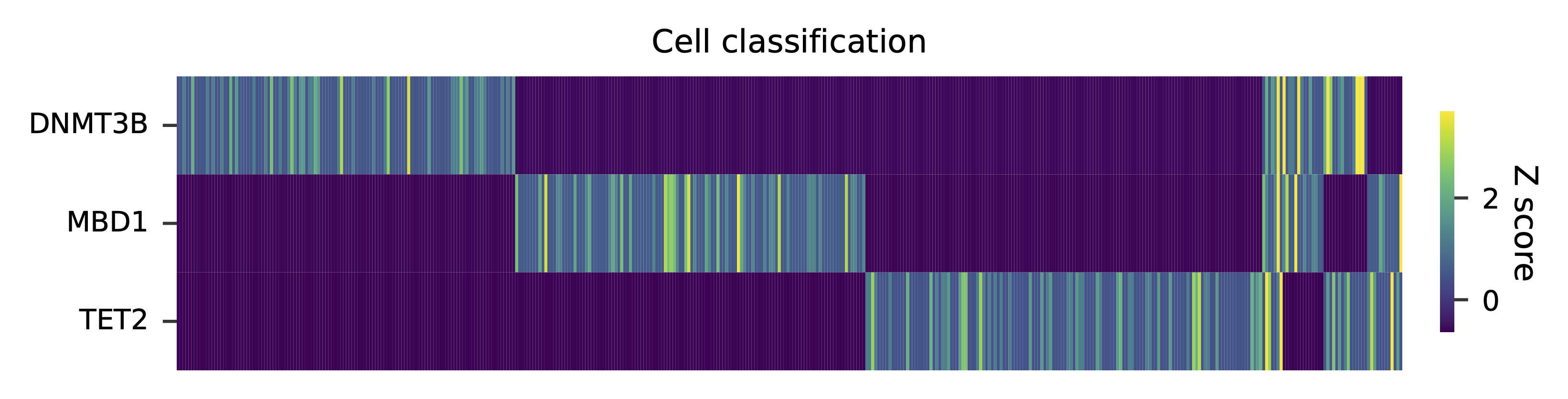
Preview the demultiplexing result: the numbers of singlets and multiplets.
In [1]: import pandas as pd
In [2]: m = pd.read_csv('demultiplexed/matrix_cell_identity.csv.gz', index_col=0)
In [3]: m.loc[:, m.sum(axis=0) == 1].sum(axis=1)
Out[3]:
DNMT3B 141
MBD1 150
TET2 158
dtype: int64
In [4]: sum(m.sum(axis=0) > 1)
Out[4]: 74
UMI distribution and knee point detection: